

Human Microbiome Project (HMP)
Human Microbiome Project (HMP)
 Science Highlights
Science Highlights
Microbiome Interagency Working Group Releases Interagency Strategy Plan for Microbiome Research
 In 2015, the Office of Science and Technology Policy (OSTP) of the White House, chartered a committee of sixteen federal agencies that fund research to complete a survey of all federally supported microbiome research over fiscal years 2012-2014. The Fast-Track Action Committee on Mapping the Microbiome (FTAC-MM) analysis included studies of microbial communities and their ecological roles in plants, animals, and humans and in ecosystems like oceans and forests. The FTAC-MM identified an investment of $920M in both intramural and extramural microbiome research over fiscal years 2012-2014; this analysis was published in Nature Microbiology (Stulberg et al. 2016). This analysis motivated the establishment of an interagency committee, the Microbiome Interagency Working Group (MIWG), which is charged with coordination of microbiome research across the federal government. This report, the ‘Interagency Strategic Plan for Microbiome Research’, released April 19, 2018, summarizes each agency’s investments in this field, the range of current coordination activities and plans for future MIWG activities to support the needed resources for advancing this emerging field. The report also includes the representative from each agency who contributed to this report.
In 2015, the Office of Science and Technology Policy (OSTP) of the White House, chartered a committee of sixteen federal agencies that fund research to complete a survey of all federally supported microbiome research over fiscal years 2012-2014. The Fast-Track Action Committee on Mapping the Microbiome (FTAC-MM) analysis included studies of microbial communities and their ecological roles in plants, animals, and humans and in ecosystems like oceans and forests. The FTAC-MM identified an investment of $920M in both intramural and extramural microbiome research over fiscal years 2012-2014; this analysis was published in Nature Microbiology (Stulberg et al. 2016). This analysis motivated the establishment of an interagency committee, the Microbiome Interagency Working Group (MIWG), which is charged with coordination of microbiome research across the federal government. This report, the ‘Interagency Strategic Plan for Microbiome Research’, released April 19, 2018, summarizes each agency’s investments in this field, the range of current coordination activities and plans for future MIWG activities to support the needed resources for advancing this emerging field. The report also includes the representative from each agency who contributed to this report.
Dramatic Fluctuations of the Gut Microbiome in Individuals with Inflammatory Bowel Disease
![]() Inflammatory Bowel Disease (IBD) is a group of conditions caused by chronic inflammation in the digestive tract. The most common subtypes of IBD are Crohn’s disease and ulcerative colitis. iHMP researchers are generating a public database containing information collected over time from several studies of IBD patients. Along with a variety of relevant patient data, the database will contain information about both the composition of the gut microbiome over time in these patients as well as the small molecules and proteins being produced by these microbes.
Inflammatory Bowel Disease (IBD) is a group of conditions caused by chronic inflammation in the digestive tract. The most common subtypes of IBD are Crohn’s disease and ulcerative colitis. iHMP researchers are generating a public database containing information collected over time from several studies of IBD patients. Along with a variety of relevant patient data, the database will contain information about both the composition of the gut microbiome over time in these patients as well as the small molecules and proteins being produced by these microbes.
It has been previously established that patients with IBD have large differences in the composition of their gut microbiome compared to heathy individuals. These findings are often based on samples obtained at a single point in time. However, the species that make up our gut microbiome can fluctuate over time based on infections, diet, antibiotics, and other factors. As part of the larger iHMP study on IBD, Dr. Jansen and colleagues determined the composition of the gut microbiomes in over 100 patients with IBD in three month intervals to determine how these microbiomes differs over time. They found that patients with IBD had much less consistency in their microbiomes when compared to healthy individuals. This information could potentially be used by doctors to monitor the health of IBD patients and their response to treatments. In addition, understanding the dynamics of the microbiome in IBD patients should help the design of future therapies that aim to restore the microbiome to a more natural state.
Watch a video describing this study.
Reference:
Dynamics of the human gut microbiome in inflammatory bowel disease. Halfvarson J, Brislawn CJ, Lamendella R, Vázquez-Baeza Y, Walters WA, Bramer LM, D'Amato M, Bonfiglio F, McDonald D, Gonzalez A, McClure EE, Dunklebarger MF, Knight R, Jansson JK. Nat Microbiol. 2017 Feb 13; 2:17004.
The genome sequence of Christensenella minuta: a bacterium which is the most heritable member of the gut microbiota and which also influences host weight.
 There has been considerable interest in a particular member of the gut microbiota, a bacterial species named Christensenella minuta, which is a member of the Firmicutes phylum. Three recent findings have generated this interest. First, the Christensenella genus appears to be found so far only in human hosts. Second, C. minuta was found to be the most heritable species in the human gut microbiome, meaning that it is the species whose presence or absence in our gut is mostly determined by the genes of its human host. Third, evidence suggests that the presence of C. minuta can directly affect the weight of its host by as yet unknown mechanisms. C. minuta is more likely to be present in leaner people, and adding C. minuta to the gut tracts of mice resulted in leaner mice. These findings suggest a potential use of C. minuta as a probiotic for weight control.
There has been considerable interest in a particular member of the gut microbiota, a bacterial species named Christensenella minuta, which is a member of the Firmicutes phylum. Three recent findings have generated this interest. First, the Christensenella genus appears to be found so far only in human hosts. Second, C. minuta was found to be the most heritable species in the human gut microbiome, meaning that it is the species whose presence or absence in our gut is mostly determined by the genes of its human host. Third, evidence suggests that the presence of C. minuta can directly affect the weight of its host by as yet unknown mechanisms. C. minuta is more likely to be present in leaner people, and adding C. minuta to the gut tracts of mice resulted in leaner mice. These findings suggest a potential use of C. minuta as a probiotic for weight control.
HMP researchers at WashU have now published the complete genome sequence of C. minuta. The availability of this genome sequence will allow future researchers to gain in depth insights into its life cycle, its metabolism and the mechanisms by which it can affect the human host in which it lives. This research highlights another way in which the HMP is supporting the larger microbiome research community.
Reference:
Genome Sequence of Christensenella minuta DSM 22607T. Rosa BA, Hallsworth-Pepin K, Martin J, Wollam A, Mitreva M. Genome Announc. 2017 Jan 12; 5(2).
The Gut Microbiome Influences Circadian Rhythms
 In yet another example of the profound role of the microbiome in regulating host physiology, a study from the HMP awardee Dr. Eugene B. Chang and colleagues examined the role of the gut microbiome in regulating the host circadian clock, control of which in mammals is located in the brain. Previous research has shown that the host circadian clock regulated microbiome composition. This study now demonstrates that the microbiome regulates the host clock.
In yet another example of the profound role of the microbiome in regulating host physiology, a study from the HMP awardee Dr. Eugene B. Chang and colleagues examined the role of the gut microbiome in regulating the host circadian clock, control of which in mammals is located in the brain. Previous research has shown that the host circadian clock regulated microbiome composition. This study now demonstrates that the microbiome regulates the host clock.
In germ-free mice they showed that the mice exhibited significant differences in their metabolism compared to conventional mice, including in their circadian clock genes. They then showed that the microbiome has its own circadian rhythm that was independent of the feeding cycle or any other cycles in the mice. Perhaps most importantly this study demonstrated that a key short chain fatty acid named butyrate was produced by the microbiome with a circadian rhythm pattern and that butyrate can directly influence the host circadian clock. Finally they provided evidence that a high fat diet could alter the microbiome circadian rhythm, thereby suggesting a link between diet, gut microbiota and obesity, all due to dysregulation of the microbiome circadian clock because of diet.
Reference:
Effects of diurnal variation of gut microbes and high fat feeding on host circadian clock function and metabolism. Leone V, Gibbons SM, Martinez K, Hutchison AL, Huang EY, Cham CM, Pierre JF, Heneghan AF, Nadimpalli A, Hubert N, Zale E, Wang Y, Huang Y, Theriault B, Dinner AR, Musch MW, Kudsk KA, Prendergast BJ, Gilbert JA, Chang EB. Cell Host Microbe. 2015 May 13, 17(5):681-9
FTAC-MM Assesses Microbiome Research in the US
 The Human Microbiome Project not only served as a catalyst for microbiome research across the National Institutes of Health (NIH), it stimulated interest in the larger growing field of microbial ecology. In the spring of 2015, the Office of Science Technology and Policy (OSTP) chartered a committee of government scientists in 14 agencies to form the Fast Track Action Committee-Mapping the Microbiome (FTAC-MM). The FTAC-MM was charged with conducting a portfolio analysis of human-, animal- and habitat-associated intramural and extramural microbiome research support over fiscal years 2012-2014. The analysis focused on the use of genome-enabled approaches to study microbial communities. It also endeavored to classify the studies into basic or applied research or tools and resource development and categorized the research into eight microbial categories and eight environments.
The Human Microbiome Project not only served as a catalyst for microbiome research across the National Institutes of Health (NIH), it stimulated interest in the larger growing field of microbial ecology. In the spring of 2015, the Office of Science Technology and Policy (OSTP) chartered a committee of government scientists in 14 agencies to form the Fast Track Action Committee-Mapping the Microbiome (FTAC-MM). The FTAC-MM was charged with conducting a portfolio analysis of human-, animal- and habitat-associated intramural and extramural microbiome research support over fiscal years 2012-2014. The analysis focused on the use of genome-enabled approaches to study microbial communities. It also endeavored to classify the studies into basic or applied research or tools and resource development and categorized the research into eight microbial categories and eight environments.
The results of the FTAC-MM analysis were published in the inaugural issue of Nature Microbiology (January 2016) in a paper titled “An Assessment of US Microbiome Research” . The analysis showed that microbiome research received a high level of support ($922M) in fiscal years 2012 -2014 across multiple federal agencies, with NIH-supporting the bulk of the research at 59%. The majority of the research was in human subjects (37%) or animal models (29%) and focused on the gut microbiome.
Other habitats that were examined included everything from studies on oceanic microbiomes (National Science Foundation) to the microbiomes of pollinators (U.S. Department of Agriculture), to the microbiomes of soils from the arctic, tropics, wetlands and grasslands (multiple agencies). About 70% of all research included in the analysis focused on total microbial community studies, which verified that the analysis captured the appropriate research.
A number of needs for the future health and growth of the field were identified, including the need for references and standards for the field, microbiome databases linking data from multiple habitats, further development of methods to study the functional properties of the microbiome and the need to train students in microbial ecology, multidisciplinary research and hypothesis-driven study design. Both the report and paper concluded with the recognition that the diverse group of governmental agencies with different missions and different constituencies arrived at the same common needs for advancing the field.
References:
An assessment of US microbiome research.![]() Stulberg E, Fravel D, Proctor L, Murray D, LoTempio J, Chrisey L, Garland J, Goodwin K, Graber J, Harris MC, Jackson S, Mishkind M, Porterfield DM, Records A. Nature Microbiology. 11 January 2016.
Stulberg E, Fravel D, Proctor L, Murray D, LoTempio J, Chrisey L, Garland J, Goodwin K, Graber J, Harris MC, Jackson S, Mishkind M, Porterfield DM, Records A. Nature Microbiology. 11 January 2016.
REPORT OF THE FAST-TRACK ACTION COMMITTEE ON MAPPING THE MICROBIOME
ASM ADVISORY ON THE REPORT OF THE FAST-TRACK ACTION COMMITTEE ON MAPPING THE MICROBIOME
A Novel Approach to Gene Sequencing Reveals Hidden Depths in Microbial Diversity
Advances in DNA sequencing technologies have been a boon for modern human microbiome studies. However, until very recently, these technologies have also had an important limitation. The most common methods have involved the extraction of DNA from these microbiomes and analysis of numerous short stretches of this DNA by sequencing. As the typical microbiome is comprised of thousands of microbial species and millions to trillions of microbial cells, it has been very difficult to re-assemble these short stretches, known as sequence reads, back into the complete genomes of these microbes. Further, with the average bacterial genome about 3,000 base pairs (bp) and the average stretch of DNA sequence read about 100-400 bps, the process of re-assembling millions of these genomes from these short reads has been very difficult.
Reassembling genomes is particularly limiting for performing metagenomics analysis which seeks to uncover the diversity of microbial communities directly from environmental samples, like from the gut tract or skin microbiomes. Finally, although the majority of microbial diversity in microbiomes is found at the subspecies and strain levels, current sequencing technologies have not been able to produce the level of detail needed to get at this level of microbial diversity.
A new study, published December 14, 2015 in Nature Biotechnology, from the laboratory of HMP awardee Dr. Michael Snyder at Stanford University, addresses this important biological problem in the microbiome field with a technical solution. The technique described in Dr. Snyder’s study, used a new sequencing technology, known as TruSeq synthetic long read sequencing technology, to dive deeper into the human gut microbiome. This technology yields 30,000-40,000 bp long reads and allows the investigators to more completely assemble whole microbial genomes from this long read sequence data. More importantly, they were able to consistently recover sufficiently long sequences that allowed them to identify sub-species and strains of bacteria and specific metabolic genes in these strains from these gut microbiome samples and thereby capture the true diversity and metabolic abilities of a microbial community.This now unmasked diversity may lead to new approaches to understanding the specific roles of these microbial strains in human health and disease.
References:
Synthetic long-read sequencing reveals intraspecies diversity in the human microbiome. Kuleshov V, Jiang C, Zhou W, Jahanbani F, Batzoglou S, Snyder M. Nature Biotechnology. 14 December 2015. 10.1038/nbt.316.
Different Treatments for Crohn’s Disease in Pediatric Patients Have Distinct Effects on the Gut Microbiome
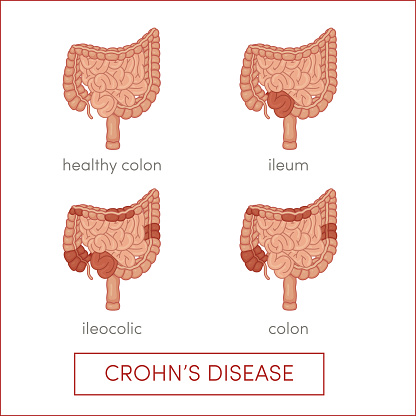 Changes in diet and application of antibiotics and/or anti-inflammatories are the typical interventions used as the standards of care for the treatment of Crohn’s disease (CD), a subtype of inflammatory bowel disease. One major characteristic of CD is an imbalance in the normal composition of the microbiota in comparison to healthy controls. A recent study from Human Microbiome Project awardee Dr. Frederic Bushman and colleagues at the University of Pennsylvania sought to systematically separate the effects of these interventions on the gut microbiomes of a cohort of pediatric CD patients.
Changes in diet and application of antibiotics and/or anti-inflammatories are the typical interventions used as the standards of care for the treatment of Crohn’s disease (CD), a subtype of inflammatory bowel disease. One major characteristic of CD is an imbalance in the normal composition of the microbiota in comparison to healthy controls. A recent study from Human Microbiome Project awardee Dr. Frederic Bushman and colleagues at the University of Pennsylvania sought to systematically separate the effects of these interventions on the gut microbiomes of a cohort of pediatric CD patients.
Each intervention independently affected the microbiome in CD patients. In particular, antibiotic use seemed to worsen dysbiosis by reducing the abundances of some microbes, increasing the abundances of fungi or both, thus aggravating the condition. Anti-inflammatories, on the other hand, reduced gut microbiota dysbiosis, thereby potentially supporting recovery from CD. Certain defined diets resulted in rapid changes in the gut microbiome suggesting diet may also be an effective treatment for CD.
Since CD patients often have higher rates of gut epithelial cell shedding and/or blood in their stool, stool samples can be sequenced to use as an early indicator of this disease, even before occult blood can be detected. This study suggests that analysis of the microbiome may lead to useful biomarkers for determining the efficacy of standard treatment for CD and for providing additional tests for early detection of CD.
References:
Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn's Disease. Lewis JD, Chen EZ, Baldassano RN, Otley AR, Griffiths AM, Lee D, Bittinger K, Bailey A, Friedman ES, Hoffmann C, Albenberg L, Sinha R, Compher C, Gilroy E, Nessel L, Grant A, Chehoud C, Li H, Wu GD, Bushman FD. Nature. 14 October 2015. 18(4): 489-500.
![]()
What is a Healthy Microbiome? New Analysis Suggests That One Size Does Not Fit All
 The typical healthy person is inhabited with trillions of microbes. To better understand the role of these organisms across our body sites, we must to catalog and analyze what organisms are there and how they interact with our own cells. A new analysis of healthy microbiomes has found that each person’s microbiome is unique. Therefore, two healthy people may have very different microbial communities but still be healthy. Strikingly, the researchers found that although unique, certain communities could be used to predict characteristics. For example, whether you were breastfed as an infant and even your level of education could be predicted based on microbial communities across varying body sites. The analysis also showed that microbial communities from varying body sites on the same individual were predictive for others. For example, gut communities could be predicted by examining the oral community, even though these communities are vastly different from each other. Taken together, this new analysis will help pave the way for future studies that can begin to use microbial communities as a basis for personalizing therapies and possibly to assess the risk for certain diseases.
The typical healthy person is inhabited with trillions of microbes. To better understand the role of these organisms across our body sites, we must to catalog and analyze what organisms are there and how they interact with our own cells. A new analysis of healthy microbiomes has found that each person’s microbiome is unique. Therefore, two healthy people may have very different microbial communities but still be healthy. Strikingly, the researchers found that although unique, certain communities could be used to predict characteristics. For example, whether you were breastfed as an infant and even your level of education could be predicted based on microbial communities across varying body sites. The analysis also showed that microbial communities from varying body sites on the same individual were predictive for others. For example, gut communities could be predicted by examining the oral community, even though these communities are vastly different from each other. Taken together, this new analysis will help pave the way for future studies that can begin to use microbial communities as a basis for personalizing therapies and possibly to assess the risk for certain diseases.
Read the University of Michigan press release here
Watch Dr. Schloss explain his research here
Reference
Ding T, Schloss PD. Dynamics and associations of microbial community types across the human body. Nature. 2014 Apr 16. PMID 24739969
![]()
Researchers Show Premature Infants Can Develop Sepsis From Gut Microbes
 A research team, supported by the Human Microbiome project, have shown for the first time that gut microbes in premature infants can cause sepsis. The team was able to use stool collected at birth from a large group of premature infants to illustrate that gut microbes, some present at birth and some that colonized later, can breach the gut to cause bloodstream infections (sepsis). The team was able to prove this by whole genome sequencing to confirm that the identical strains were in both the gut and the stool. These findings are highly relevant because physicians may be able to use this information to establish a risk early, potentially remove the bacteria with treatments, and be able to increase hygiene to minimize the risk.
A research team, supported by the Human Microbiome project, have shown for the first time that gut microbes in premature infants can cause sepsis. The team was able to use stool collected at birth from a large group of premature infants to illustrate that gut microbes, some present at birth and some that colonized later, can breach the gut to cause bloodstream infections (sepsis). The team was able to prove this by whole genome sequencing to confirm that the identical strains were in both the gut and the stool. These findings are highly relevant because physicians may be able to use this information to establish a risk early, potentially remove the bacteria with treatments, and be able to increase hygiene to minimize the risk.
Read more about sepsis here
Reference
MA Carl et al. Sepsis from the gut: The enteric habitat of bacteria that cause late-onset neonatal bloodstream infections. Clinical Infectious Diseases DOI: 10.1093/cid/ciu084 (2014).
NIH Researchers Conduct First Survey of Fungal Diversity on Human Skin
A research team at the NIH, funded in part through the Common Fund Human Microbiome Project, have sequenced and analyzed the DNA of fungi that inhabit skin sites of healthy adults in order to define populations across the skin. This work yields insights that will pave the way for studies to examine the role fungi on the skin play in maintaining health and also how associated factors may contribute to the formation of skin conditions. Of the sites examined, the feet were found to be the home of the most diverse and complex sites. This study has been published in the May 22, 2013 early online issue of Nature.
For more information read the NIH press release here
March 18, 2013
New findings lead scientists to a better understanding of an elusive microbe and oral health
A team of scientists at the Oak Ridge National Laboratory (ORNL) funded by the NIH Common Fund Human Microbiome Project (HMP) have made new discoveries about a microbe that is important in human oral health. Using cutting-edge technology, the team was able to complete full sequencing of the genome from a single cell. The ability to isolate just a single bacterial cell and sequence the genome is an important component of examining the human microbiome because it allows for the study of species that cannot be cultured in the lab. The organism the examined is most closely related to sulfate reducers, which are normally found in salt marshes, sewer pipes, hot springs, and surprisingly the human mouth. Studies have shown that this type of bacteria is elevated in patients suffering from periodontitis, a disease marked by swelling and infection of areas that support our teeth. New findings presented in the current study show that this species uses a unique coding scheme that likely allows it to successfully compete in the complex oral microbial environment. The technology advancement and scientific findings reported in this study will increase our understanding of the role that our microbes play in oral health.
Reference:
Campbell JH, O'Donoghue P, Campbell AG, Schwientek P, Sczyrba A, Woyke T, Söll D, Podar M. UGA is an additional glycine codon in uncultured SR1 bacteria from the human microbiota. Proc Natl Acad Sci USA 2013, Mar 18. PMID: 23509275.
June 13, 2012
NIH Human Microbiome Project Completes Seminal Study of Microbial Diversity in Healthy Volunteers
The NIH Common Fund’s Human Microbiome Project (HMP) has just published two seminal papers in the June 14, 2012 issue of Nature and a series of additional papers in several PLoS journals (click here for more), the NIH announces on June 13, 2012. These milestone studies are centered on the project’s ambitious and unparalleled examination and analysis of the microbiomes of a healthy cohort consisting of over 240 individuals. The resources and resulting analysis shed light onto the intricate details of the complete healthy human microbiome and pave the way for future studies in the field. The diversity both within and among body sites highlights an important and complex association between humans and associated microbes.
A comprehensive community resource
One of the two Nature papers from the June 14 issue examined a population of 242 healthy adults, each of whom were sampled at 15 (male) to 18 (female) body sites, with each person sampled on one to three distinct occasions. This unparalleled effort led to DNA sequencing of microbial eukaryotes, archaea, bacteria, and viruses (both mammalian and bacterial). Using standardized protocols and methods across the four sequencing centers, the consortium was able to generate 5,177 unique microbial taxonomic profiles (from 16S rRNA gene sequences) and over 3.5 Tbp of metagenomic sequence. Furthermore, their studies led to the assembly of hundreds of reference genomes from the human microbiome. These efforts have led to an expansive generation of genomic data and also extensive data related to functional proteins and site-specific metabolism. The targeted approach of assembling data in a site-specific manner allowed the researchers to assemble less abundant organisms that were common across the cohort. In addition to the microbial analyses, healthy cohort subjects also submitted blood samples so that human genome analysis and cell-line development can be implemented in future studies. This foresight in the project’s planning unlocks an area of great potential for benefits to human health. Much of the data, other than protected health information, is publicly available via NCBI HMP project page and the HMP Data Analysis and Coordinating Center (DACC).
Extensive analysis of the healthy human microbiome
After establishing standards for data generation, the HMP consortium continued on to conduct a comprehensive analysis of the largest human cohort and set of distinct, clinically relevant body habitats to date (five major habitats). This is the first study to include metagenomic data (data that does not rely on culturing microbes) across body habitats from a cohort of this magnitude, in an attempt to describe the basics of overall host associated microbial life as well as the basics of microbial life for each host site examined. The research team found that there was strong site specialization both within and among subjects but that the diversity and abundance of each habitat’s signature microbes varied widely among the healthy subjects. Somewhat surprisingly based on the genetic sequence with large phylogenetic variations and general variation among the individual samples, there was remarkable functional stability. In essence, the authors illustrate that while the compositions vary widely the functionality is similar, meaning that there are many ways to construct microbial communities to perform similar functions.
Through this analysis, the consortium was also able to make general characterizations about the human microbiome. One finding was a limited, but commonly detectable, number of pathogens, leading to speculation that a low abundance of potentially harmful microbes might in some cases be beneficial to the host. Another interesting finding was patterns of alpha and beta diversity, where alpha diversity is defined as the diversity within a site and beta diversity is defined as that observed among subjects. For example, saliva was shown to have high alpha diversity (many different taxonomical units) but low beta diversity (very similar among the cohort). Human sites varied widely in alpha and beta diversity and future characterizations of the microbiome and its relation to human diseases will likely shed further light onto the importance of these variations in healthy and disease states.
A major finding from the analysis of the healthy cohort was a number of well-validated correlations of taxa (groups of organisms) and function with host phenotypes. Some of the greatest correlations observed were between ethnicity and microbiome composition across all body habitats and a positive correlation of vaginal pH to microbial diversity (higher pH having higher diversity). Furthermore, there was an intriguing association of age with skin microbiome-associated metabolic pathways and oral microbiome composition, and a modest correlation between microbial composition and body mass index. Overall, many correlations were observed but as of now most of the data is not fully understood and requires future studies and examinations of additional factors including diet and host genetics.
A true team effort
The results presented in these papers highlight a remarkable level of collaboration among a large number of researchers. Interactions and collaborations among the two clinical centers and four sequencing centers were paramount for success. During the early stages of the program, data were being generated at an exponentially faster rate than analyses could be performed. To address these issues, the consortium formed the Data Analysis Working Group (DAWG), which consists of members from the genome centers and computational tools groups in addition to several experts not directly supported by the HMP. This was critical for the success of this large-scale and collaborative process. The partnerships and synergism from this teamwork will continue to fuel microbiome research.
The two landmark papers and the series of companion papers establish a foundation to catalyze and aid a myriad of studies ranging from basic to translational to clinical. For more information about the NIH Common Fund Human Microbiome Project please visit the Common Fund HMP and HMP Data Analysis and Coordinating Center (DACC) websites.
A highly adapted genome: Sequence of immune-regulating bacteria reveals why culturing attempts have been unsuccessful
 A team of researchers, funded in part by the NIH Common Fund’s Human Microbiome Project, have sequenced and analyzed a class of unique bacteria that has eluded growth in the lab setting for over forty years. These segmented filamentous bacteria (SFB) are found in mice and other mammals and are known as the first commensal (non-pathogenic) bacteria identified that affect the host immune system. In the current study, researchers collected droppings from mice that were only colonized with SFB and used next generation sequencing platforms to obtain the sequence and construct the complete genome. They found that the genome was much smaller than closely related species and similar to other “minimal” bacteria that have been studied. This was due to a lack of many genes related to metabolism. It appears that much of the genetic material was lost because the bacteria rely on the host for a great deal of what they need to grow and survive. In fact, one of the few classes of genes in abundance are those related to transport of metabolites from the environment (host gut). These findings explain why is has been so difficult to grow these organisms outside of the host and highlights the close association of these bacteria with their host. This incredibly close association between host and microbe could be one reason as to why these bacteria help recruit immune cells that protect their host from pathogenic enteric bacteria. Although SFB have yet to be discovered in humans, the findings from this study will be an important resource for further examination of the role microbes play in host immune systems and overall metabolism.
A team of researchers, funded in part by the NIH Common Fund’s Human Microbiome Project, have sequenced and analyzed a class of unique bacteria that has eluded growth in the lab setting for over forty years. These segmented filamentous bacteria (SFB) are found in mice and other mammals and are known as the first commensal (non-pathogenic) bacteria identified that affect the host immune system. In the current study, researchers collected droppings from mice that were only colonized with SFB and used next generation sequencing platforms to obtain the sequence and construct the complete genome. They found that the genome was much smaller than closely related species and similar to other “minimal” bacteria that have been studied. This was due to a lack of many genes related to metabolism. It appears that much of the genetic material was lost because the bacteria rely on the host for a great deal of what they need to grow and survive. In fact, one of the few classes of genes in abundance are those related to transport of metabolites from the environment (host gut). These findings explain why is has been so difficult to grow these organisms outside of the host and highlights the close association of these bacteria with their host. This incredibly close association between host and microbe could be one reason as to why these bacteria help recruit immune cells that protect their host from pathogenic enteric bacteria. Although SFB have yet to be discovered in humans, the findings from this study will be an important resource for further examination of the role microbes play in host immune systems and overall metabolism.
Reference:
Sczesnak A, Segata N, Qin X, Gevers D, Petrosino JF, Huttenhower C, Littman DR, Ivanov II. The genome of th17 cell-inducing segmented filamentous bacteria reveals extensive auxotrophy and adaptations to the intestinal environment. Cell Host Microbe. 2011 Sep 15;10(3):260-72. PMID 21925113.
The Human Microbiome Project in 2011 and Beyond
Dr. Lita Proctor, coordinator for the Human Microbiome Project (HMP), National Human Genome Research Institute, gives an overview of the HMP program and describes the vast resources produced thus far from the unprecedented study of 300 healthy individuals. Ongoing studies of specific diseases (demonstration projects) and the future directions of human microbiome research are also discussed.
Reference:
Cell Host Microbe. 2011 Oct 4; 10(4): 287-91
Getting Personal with Bacteria
Microbes, including bacteria, inhabit your body in great numbers and impact many aspects of health and disease such as obesity and Crohn's disease. Characterizing the genetic diversity of microbes that live in specific areas of the body is key to understanding the composition and dynamics of microbial communities within individuals, in transmission between individuals, and in transmission between individuals and the environment. The ability to characterize microbial diversity and transmission has been hampered in the past by a lack of high-throughput analysis tools. New computational tools being developed through the Common Fund's Human Microbiome Project (HMP) are accelerating microbiology and biomedical research, and unexpectedly, other fields like forensics.
Dr. Rob Knight, an investigator in the HMP, is developing novel approaches to analyze human microbial communities, and recently contributed to a paper in the Proceedings of the National Academy of Science on the discovery of "microbial fingerprints"; in a person's skin. The skin surface harbors a large number of bacteria that are highly diverse and yet personally unique from individual to individual. The bacteria are easily dislodged from the skin and transferred to objects upon contacting. By analyzing the "microbial fingerprint"; of bacteria left on computer equipment, Dr. Knight and colleagues at the University of Colorado found that the fingerprint could be traced to a specific individual with a high degree of certainty even if the objects had not been touched for two weeks. The approach could be important in forensic investigations to provide independent confirmation of forensic results obtained using more traditional methods such as human DNA analysis or fingerprinting.
